Why PXR Matters
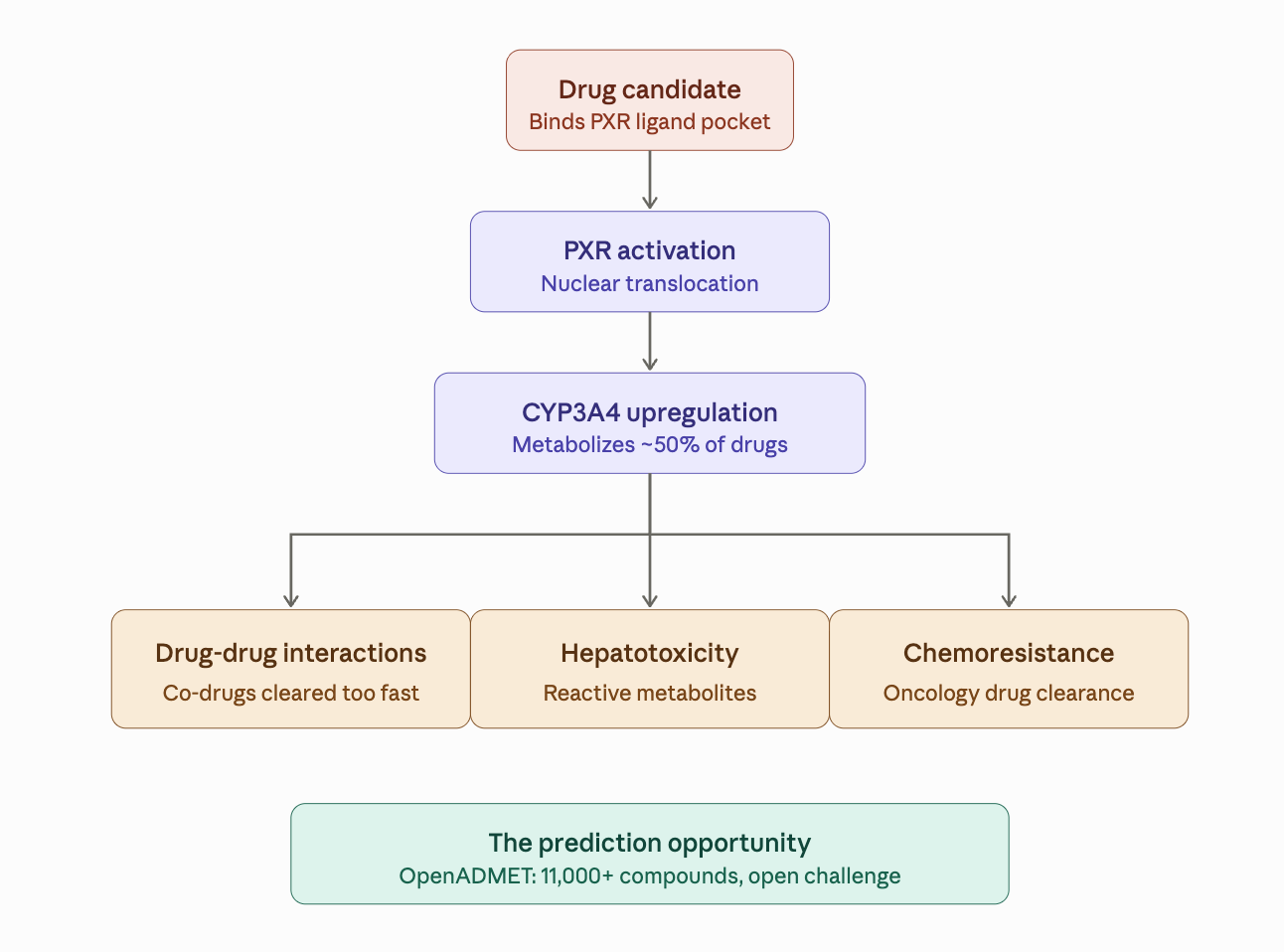
The pregnane X receptor, or PXR, is a nuclear hormone receptor that sits in the cell like a smoke alarm for xenobiotics. When a foreign molecule (including many drug candidates) binds to PXR’s ligand-binding domain, PXR moves to the nucleus and activates the transcription of drug-metabolizing enzymes — most critically CYP3A4, the enzyme responsible for metabolizing approximately 50% of all marketed drugs.
This is bad news for drug discovery programs in three main ways:
Drug-drug interactions. If your compound activates PXR, it will accelerate the metabolism of any co-administered drug that’s also a CYP3A4 substrate. That can reduce the co-administered drug’s plasma concentration below therapeutic levels, causing clinical failures that have nothing to do with the primary drug’s target.
Hepatotoxicity. Upregulated CYP3A4 means more reactive metabolites. That raises the risk of liver toxicity through covalent modification of hepatic proteins.
Chemoresistance. In oncology, PXR activation in tumor cells can enhance the clearance of chemotherapeutic agents, making cancer drugs less effective.
The challenge with predicting PXR induction is that the receptor has an enormous, flexible binding pocket — on the order of 1,200 to 1,600 cubic angstroms. It accommodates ligands across an extraordinary range of sizes and shapes. Traditional pharmacophore-based methods struggle because there isn’t one pharmacophore; there are many.
Until recently, the public data has been sparse. ChEMBL contains about 800 high-quality PXR pEC50 values scattered across 150 papers. That’s not enough for modern machine learning approaches to reach their potential.
The OpenADMET PXR Challenge
OpenADMET, the open-science consortium building predictive models for ADMET properties, has just opened a blind challenge to predict PXR induction. In partnership with Octant (who generated the assay data) and the Fraser Lab at UCSF (who contributed structural biology capabilities), they’ve released the largest public PXR dataset ever assembled: over 11,000 compounds run through a rigorous assay cascade.
The cascade is worth understanding because it shapes how the data should be used:
- Primary screen: 11,362 diverse compounds screened at a single concentration
- Dose-response curves: 4,779 compounds advanced to 8-point DRCs
- Refinement: 114 compounds with EC50 ≤ 1 µM
- Counter-screen: A PXR-null cell line filters out non-specific activators, yielding 63 potent, selective agonists
- Analog expansion: Similarity searches of the 63 actives produced the 513-compound test set
What makes this dataset special is step 4 — the counter-screen. The assay uses a chimeric reporter where PXR’s ligand-binding domain drives luciferase expression. A parallel counter-assay uses the same system but with PXR mutations that abolish function. If a compound lights up both assays, it’s a false positive — probably a general transcriptional activator, HDAC inhibitor, or assay interference. This counter-screen data is included in the training set. OpenADMET’s own internal testing suggests that using this data well is the key to strong model performance.
The challenge runs in two phases:
- Phase 1 (through May 25): Participants predict pEC50 for all 513 test compounds; the live leaderboard reflects performance on “Analog Set 1.”
- Phase 2 (May 26 through July 1): Analog Set 1 values are unblinded. Participants refine their predictions for Analog Set 2, which remains fully blinded until the end.
This design simulates the iterative nature of real drug discovery, where new data arrives and you have to incorporate it on the fly.
Why I’m Entering
I started Scriptome.AI because I believe the biggest bottleneck in applying AI to drug discovery isn’t the AI itself. It’s the gap between AI tools and the scientists who understand the biology. Most small biotechs have brilliant domain experts who don’t yet have the ML fluency to evaluate which tools actually work. And most ML practitioners don’t have enough context to know which problems are actually worth solving.
The PXR challenge sits right in that gap. It’s not a generic benchmark. It’s a problem that has derailed real drug discovery programs, with data shaped by the same kind of assay cascade you’d see at a biotech company. Solving it well requires both ML competence and medicinal chemistry intuition.
I’m not a computational chemistry specialist. I’ve spent 15 years in drug development roles at Sanofi Genzyme, Hopewell Therapeutics, and NanoString Technologies. I have degrees in chemical engineering, biochemistry, and an MBA in business analytics. Over the last two years, I’ve been aggressively upskilling in AI/ML, co-teaching a course called The Fast and the Curious with Jesse Johnson of Merelogic to help other bench scientists do the same.
This challenge is, in a real sense, the final exam for everything I’ve been teaching. And I think there’s value in failing or succeeding in public.
Building in Public
Over the next 10 weeks, I’ll be documenting this journey in detail. The plan:
- Weekly LinkedIn posts covering each stage of the work
- Longer-form blog posts here at Scriptome.AI with code, data visualizations, and lessons
- Two Scriptome Podcast episodes: one early on the fundamentals, one mid-challenge retrospective
- A final wrap-up after Phase 2 closes on July 1
I’ll share what works and what doesn’t. I’ll show the mess of real ML work, not just the polished end result. And I’ll try to translate between the ML and domain-expert perspectives throughout.
My technical approach starts simple and gets progressively more ambitious:
- Baseline: LightGBM on Morgan fingerprints plus RDKit physicochemical descriptors
- Feature engineering: Exploit the counter-assay selectivity data
- Multi-task learning: Predict primary pEC50, counter-assay pEC50, and Emax jointly
- Ensembles: Combine LightGBM, XGBoost, and neural network predictions with uncertainty estimates
- Advanced representations: Incorporate learned molecular embeddings (ChemBERTa, Uni-Mol) and possibly graph neural networks
- Phase 2 pivot: Retrain with the unblinded Analog Set 1 data and focus on activity cliff analysis
None of this is exotic by ML standards. The differentiator — if there is one — will come from using domain knowledge to engineer features and interpret results that pure ML approaches might miss.
Looking for Teammates
Here’s where I could use help.
This challenge rewards cross-functional thinking. The organizers have hinted strongly that using the counter-assay data well is the key to performance — and that’s as much an assay design and medicinal chemistry question as it is a machine learning one.
I’m assembling a small, cross-functional team to work on this together. I’d love to hear from:
- Computational chemists or ML practitioners who want to pair modern techniques with deep domain context
- Medicinal chemists who want to think through activity cliffs, scaffold families, and SAR patterns
- Anyone with experience in graph neural networks, multi-task learning, or learned molecular representations
- Anyone curious who wants to learn alongside us
The structure would be flexible: shared alias on the leaderboard, joint credit on the required methodology report, a weekly sync, and asynchronous collaboration otherwise. We can submit anonymously if that’s preferred.
If you’re interested, reach out to me on LinkedIn or email stu@scriptome.ai.
What I Hope to Learn
More important than my final leaderboard position is what this experience will teach me — and, through these posts, teach the broader community — about the current state of ADMET prediction.
Can modern ML actually solve PXR induction prediction? The honest answer today is: we don’t know. OpenADMET’s previous challenge on ExpansionRx data showed that ML models have made real progress on ADMET endpoints, but also revealed significant gaps. PXR is particularly hard because of its flexible binding pocket and because the test set is deliberately populated with activity cliffs — close structural analogs with divergent activities.
If I — or anyone — builds a model that performs well on this challenge, that’s meaningful evidence that the field has matured to the point where these predictions are usable for early triage in small biotech workflows. If no one performs well, that’s equally meaningful: it tells us we still need experimental validation and can’t skip ADMET profiling yet.
Either outcome advances the field. That’s the point of open challenges.
If you want to follow along, connect with me on LinkedIn or subscribe to The Scriptome Podcast. If you’re a biotech professional looking to build AI/ML fluency, check out The Fast and the Curious, the AI upskilling course for bench scientists I co-teach with Jesse Johnson.
The OpenADMET PXR Challenge runs through July 1, 2026. Learn more at openadmet.ghost.io and join the conversation on the OpenADMET Discord.
About the author: Stu Angus is the founder of Scriptome.AI, a Cambridge, MA-based consulting firm helping small biotech companies evaluate and adopt AI/ML tools for drug discovery. He has over 15 years of drug development experience across Sanofi Genzyme, Hopewell Therapeutics, and NanoString Technologies. He organizes the AI x Biotech Professionals Working Group and co-teaches The Fast and the Curious with Jesse Johnson of Merelogic.